近日,化工学院绿色制药与智能化工团队在机器学习势函数加速有机合成反应过渡态搜索方向取得新进展。相关成果以“Reactive machine learning potential for accelerating transition state search in organic synthesis”为题,在Nature Communications期刊发表研究论文。该论文的第一作者为大连理工大学化工学院任凯派博士生和唐坤博士生,通讯作者为刘奇磊副教授。

有机合成反应的动力学行为往往受控于过渡态结构及其对应的反应能垒,精准表征过渡态,对于深入揭示反应速率、选择性与反应机理至关重要。然而,传统密度泛函理论(DFT)计算成本高,难以支撑大规模反应网络与复杂药物化学反应的高通量研究;半经验量子化学方法虽具备更高效率,但在过渡态结构与能量预测方面精度有限。如何在“DFT精度”与“高通量效率”之间取得平衡,成为计算有机化学与智能合成领域的核心挑战。针对这一问题,研究团队提出了面向有机合成的反应机器学习势模型DeePEST-OS(Deep learning-based molecular Potential Energy Surface prediction Tool for Organic Synthesis)。该模型融合半经验量子化学方法GFN2-xTB的物理先验知识与MACE高阶等变消息传递神经网络,并通过Δ-learning策略学习DFT与GFN2-xTB之间的系统偏差,在显著提升计算效率(相比DFT加速约10,000倍)的同时,实现接近DFT精度的过渡态搜索与反应能垒预测。
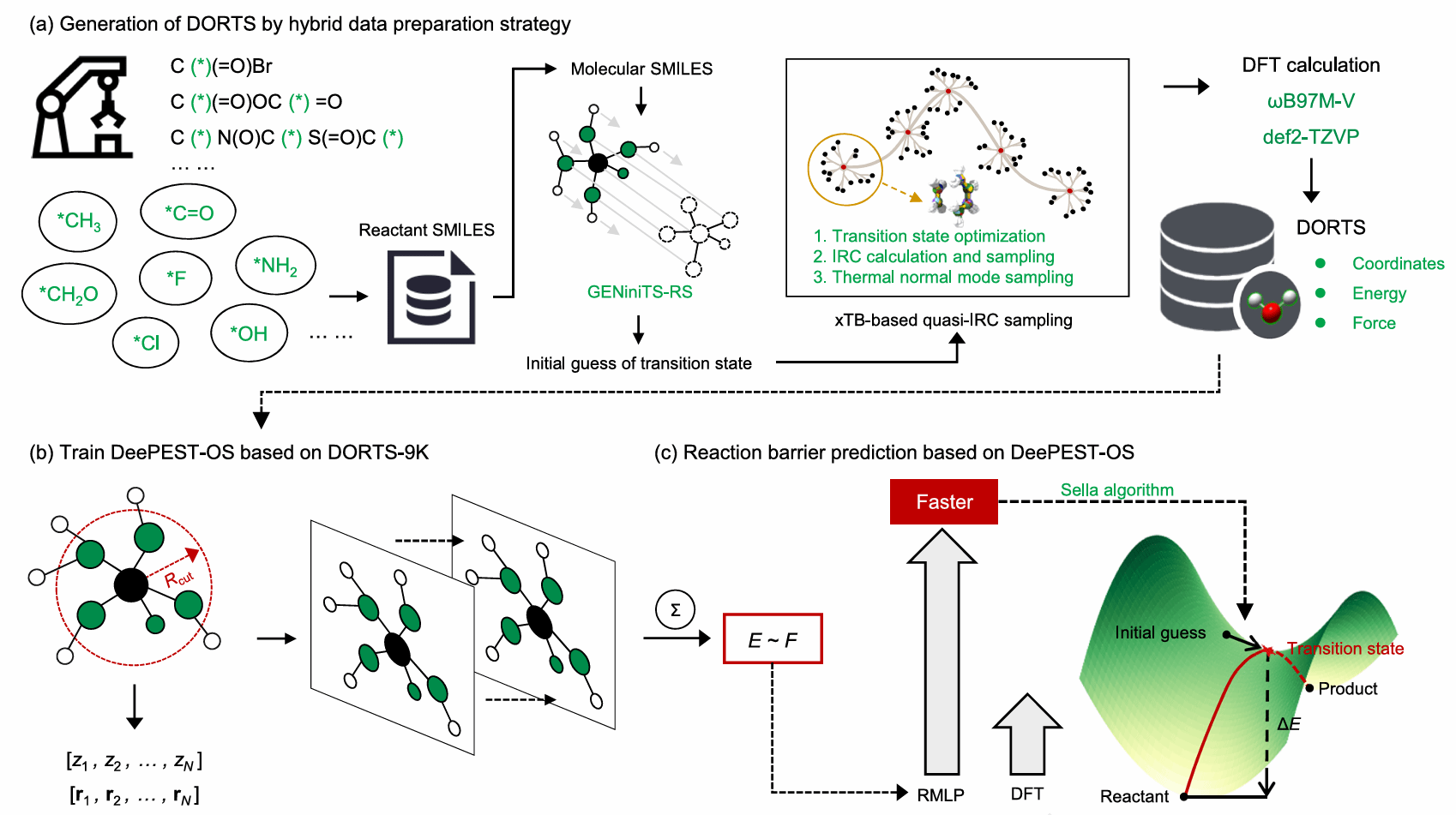
图1 DeePEST-OS建模方法
本研究首先构建了一个全新的海量有机反应过渡态数据库DORTS(Database of Organic Reaction Transition States)。研究团队从《March’s Advanced Organic Chemistry》中手动提取了255类基础有机反应模板,结合计算机辅助分子设计方法、过渡态初猜自动生成、基于GFN2-xTB的准反应路径采样、简正模采样方法和高精度DFT标注,获得了74,837条有机反应路径和约750万个反应路径构象。与常用Transition1x数据库相比,DORTS不仅反应数量更多、反应类型更丰富,还覆盖C、H、O、N、P、S、F、Cl、Br、I共10种元素,更贴近含硫、含磷和卤代有机分子等真实合成体系。
表1 DORTS数据库对比Transition1x数据库
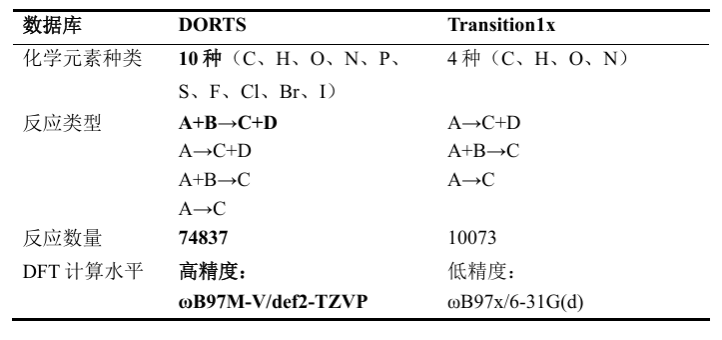

图2 DORTS数据库
在模型性能方面,DeePEST-OS在测试集中的能量预测平均绝对误差MAE仅为0.266 kcal/mol、原子力MAE为0.380 kcal/(mol·Å)。在未参与训练的DORTS-1K外部测试中,模型预测的过渡态几何结构平均均方根偏差RMSD为0.12 Å,反应能垒MAE为0.60 kcal/mol,低于1 kcal/mol的化学精度经验阈值。以苯与亚硝酸的反应为代表性测试,DeePEST-OS在过渡态优化中相较DFT方法实现近10,000倍加速。
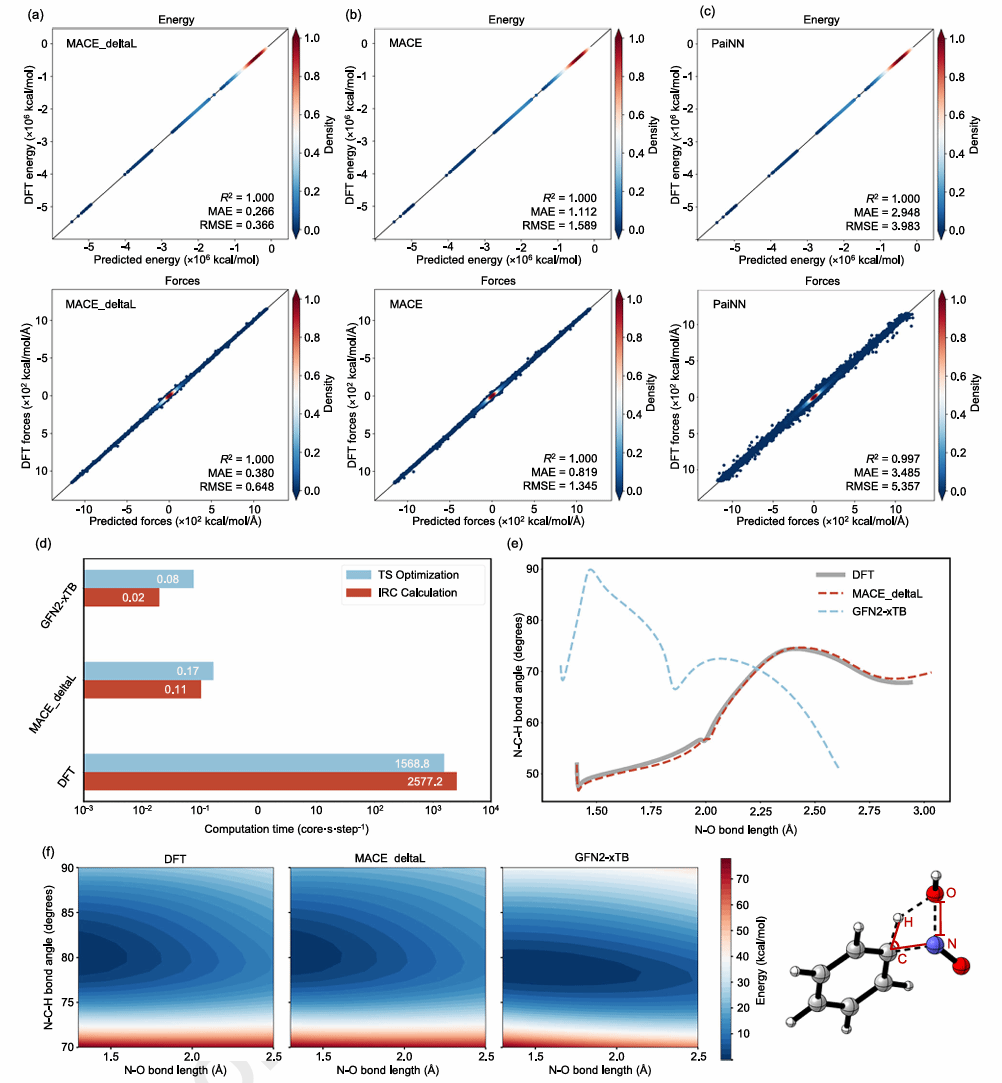
图3 DeePEST-OS模型预测精度与效率
为进一步验证模型的可迁移性,研究团队在Transition1x数据集上开展跨数据集评估。与近期提出的有机反应过渡态端到端生成模型React-OT相比,DeePEST-OS-T1x在995个成功优化的外部测试反应中表现出更低的过渡态几何RMSD(0.050 Å vs. 0.077 Å)和更低的反应能垒误差(0.692 kcal/mol vs. 1.038 kcal/mol),同时过渡态优化平均耗时为1.4秒/反应。不同于只生成几何结构的模型,DeePEST-OS在优化过渡态的同时还可以输出接近DFT水平的能量信息,更适合反应动力学分析。
DeePEST-OS还被用于复杂有机反应中的过渡态构象筛选。研究团队将其与前期开发的GENConf-TS算法结合,在10个复杂反应中筛选最稳定过渡态构象。结果显示,DeePEST-OS能够与DFT一致地识别最稳定构象,并显著降低计算成本,为研究立体选择性和构象效应提供了高效工具。
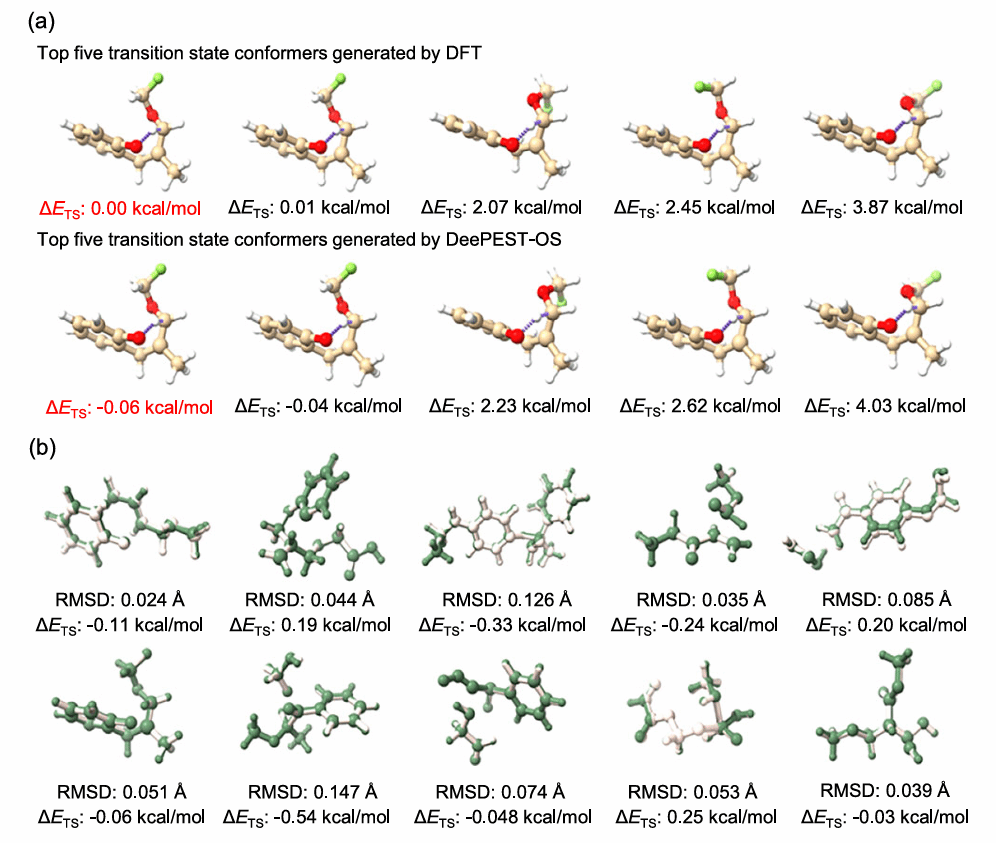
图4 能量最小过渡态构象搜索
在药物分子逆合成路线分析中,研究团队以扎托司琼(Zatosetron)为例,将DeePEST-OS与团队自主研发的逆合成规划软件RetroSynX、过渡态初猜生成方法GENiniTS-RS结合,预测多步合成路径的反应势能面。相较GFN2-xTB方法,DeePEST-OS对中间体和过渡态几何结构的平均RMSD降至0.16 Å,对反应吉布斯自由能垒的MAE降至1.18 kcal/mol。值得注意的是,相关测试中包含训练数据中未出现的结构,说明该模型并非简单记忆训练样本,而是能够学习反应路径中的原子相互作用规律,实现高精度的跨反应体系外推预测。
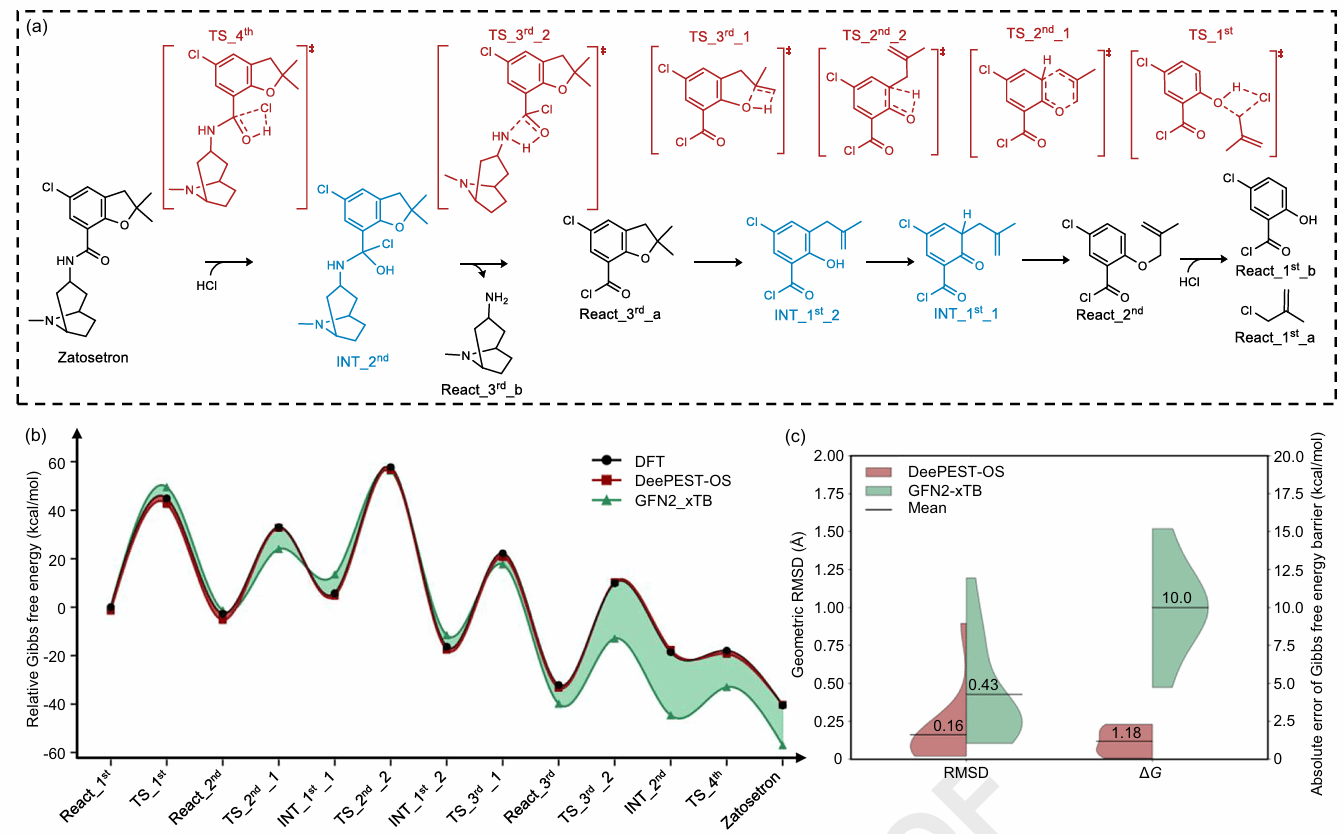
图5 DeePEST-OS应用于Zatosetron药物逆合成路线反应势能面预测
针对模型预测能否对应真实实验结果的问题,研究团队开展了Diels-Alder反应endo/exo非对映选择性的实验验证。结果表明,DeePEST-OS预测的反应吉布斯自由能垒差(ΔΔG)与实验非对映体过量值(de)具有较好的相关性,R²达0.76;对应的DFT基准方法的R²为0.82。这说明DeePEST-OS不仅可以复现DFT预测结果,也有潜力服务于面向实验的选择性预测。
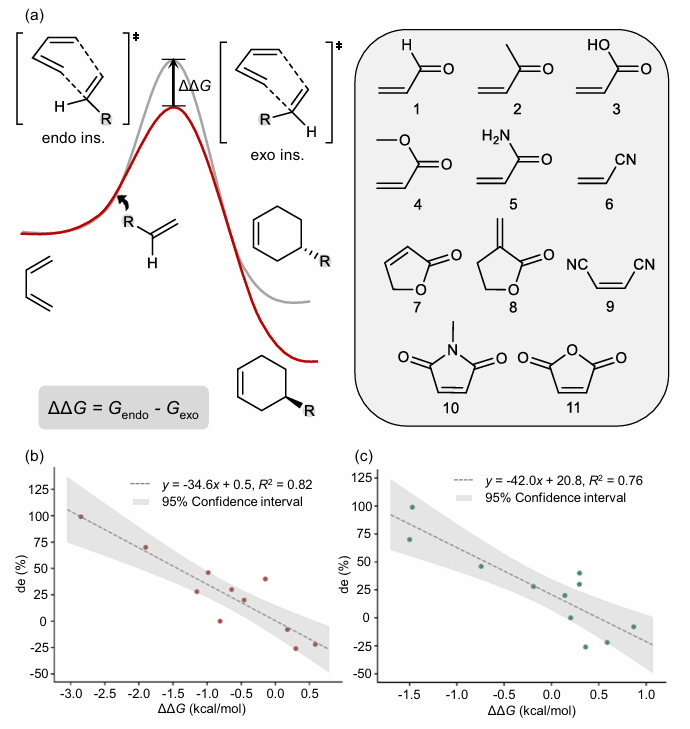
图6 Diels-Alder反应中DeePEST-OS与DFT预测的反应吉布斯自由能垒差(ΔΔG)与实验非对映体过量值(de)的线性拟合
该研究建立了从反应数据构建、机器学习势函数训练,到过渡态搜索、反应能垒预测和实验选择性验证的完整流程。所提出的DeePEST-OS反应机器学习势函数模型为多元素有机反应体系提供了一种兼顾精度与效率的计算工具,可用于药物合成路线评估和有机反应选择性预测,推动计算有机化学朝着量子精度-高通量筛选协同范式发展,最终建立一个面向实验的有机合成设计智能预测框架。
DOI: 10.1038/s41467-026-72945-0
Early-access: https://www.nature.com/articles/s41467-026-72945-0
